Gromacs system preparation
This protocol prepares a Gromacs MD system prior to its simulation from a AtomStruct object. This protein must have all the atoms including hydrogens, we recommend you to prepare it first with any of our integrated protocols for receptor preparation.
It allows the user to create a solute boundary box, define the force field and finally specify the ions in the solute, which can be set to neutralize the charges, or manually add the desired number.
Note
Unfortunately, as for today we do not include the functionality for preparing complexes containing non-protein atoms, such as ligands. Hopefully it will be coming soon.
Input
Note
All parameters include a help button that gives further information for each of them.
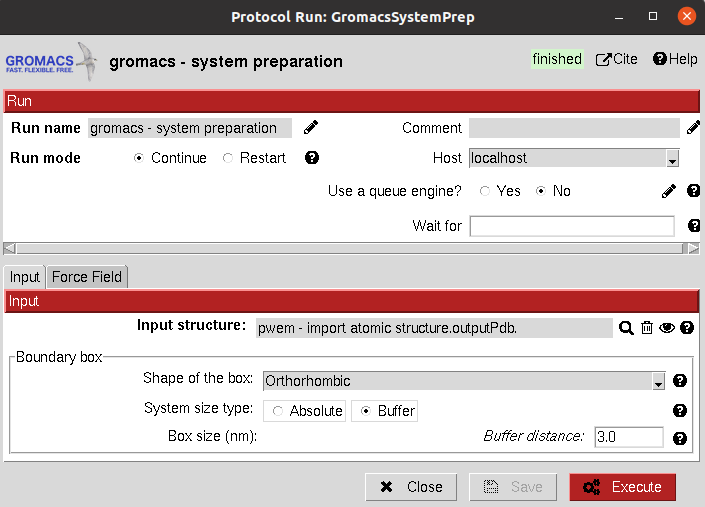
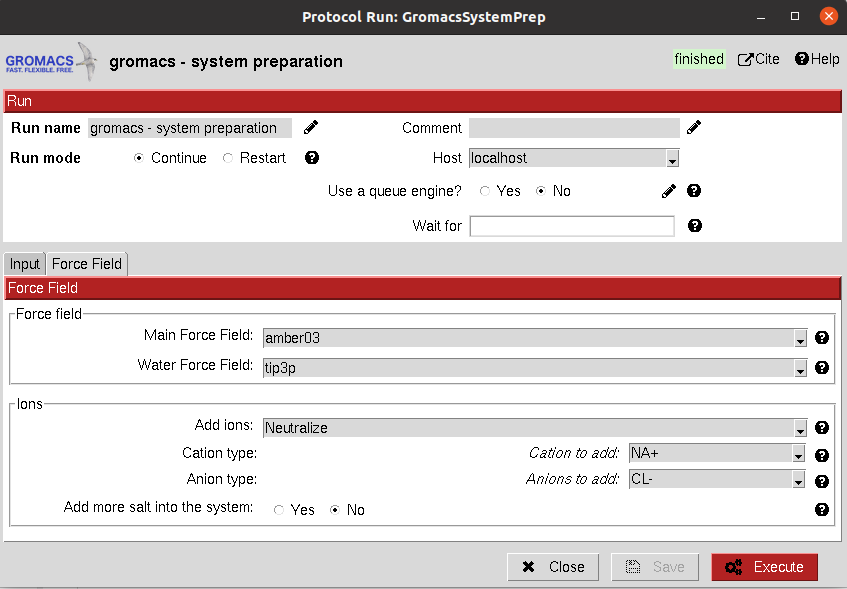
The result of this protocol is a GromacsSystem, containing the Gromacs coordinates and topology files. The user
can visualize the complex with PyMol using Analyze Results.

Test
This protocol has an integrated test that can be run using the following command:
scipion3 tests gromacs.tests.tests.TestGromacsPrepareSystem