Welcome to Scipion Chem’s documentation!
In the following documents, you will find all the necessary information for installing and using scipion-chem and all the related plugins (called scipion-chem-*). These plugins integrate mainly software for Virtual Drug Screening (VDS). You can find the complete list in https://github.com/scipion-chem
Scipion Chem is itself built as a plugin of Scipion, a workflow engine mainly used for Cryo-Electron Microscopy. Therefore, most of its documentation also applies to Scipion-chem and you will need to install Scipion3 first in order to use Scipion-chem.
Installation
Once Scipion is installed in your computer, you can install Scipion-chem as its plugin. Scipion-chem is the core for the rest of scipion-chem-* plugins, so you would need to install it first. To do so, you can check the instructions in the scipion-chem README
Then, you are free to install any subsequent plugin you are interested in. The procedure is very similar, you will find README files in each of the plugin repositories in case of doubt, appart from a more exhaustive documentation within this pages.
Scipion Chem overview
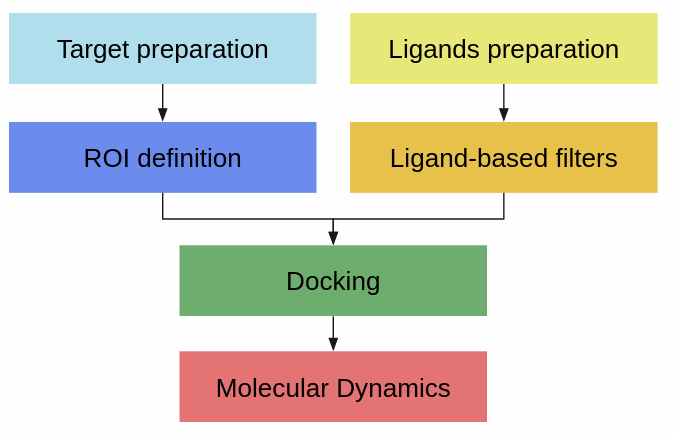
In this section, each of the Scipion Chem plugins will be briefly presented, grouped by their main functionalities. We have mainly followed the workflow shown in the figure below, even though some included tools fall outside these groups. Further documentation will be written for each of them, together with case studies and workflow examples.
1) scipion-chem
As stated before, scipion-chem is the core of the Scipion Chem plugins and it includes several tools which are mainly oriented to virtual Drug Screening.
scipion-chem automatically installs several util third-party software for the management and visualization of the tasks in a typical bioinformatics and VDS workflow. These include:
OpenBabel and RDKit: the main small molecule handlers and converters.
MGLTools: utadditional utils for small molecules, docking, … (includes AutoDockTools).
JChemPaint: Java program to manually draw small molecules.
Pymol: main viewer of Scipion Chem for small molecules and structures.
VMD: secondary viewer of Scipion-Chem for structures and Molecular Dynamics.
AliView: main viewer for sequences.
Last but not least, Scipion-chem includes 3 groups of protocols: General, Sequences and VirtualDrugScreening. As their names suggest, they contain Scipion protocols related to general or management tasks, protocols for the management of sequences and finally those mainly used in a VDS workflow, including both structure and ligand based approaches. Among them, we include consensus protocols for structural regions of interest (ROIs), which are usually predicted protein pockets, and for docking results. Further information about all these protocols can be found in the subsequent documentation.
2) Target and ligand preparation
The protocols for the protein (target) and the small molecule (ligand) preparation are actually included in other plugins and do not hold any plugin by themselves. As you will be able to read below, protocols for receptor preparation can be performed with autodock, rosetta or lephar plugins; while protocols for ligand preparation can be performed using autodock, openbabel and rdkit (these last two included in Scipion-chem).
3) Structural Regions Of Interest (ROIs)
Usually, the VDS workflow from the target starts by defining onto the structure those regions where we are interested in exploring the druggability. This is, defining those places where we would like to perform the docking.
We can address this task from multiple angles and Scipion-chem tries to offer a flexible environment to do so:
- Manually define structural ROIs: If we have some prior knowledge about the receptor, we can just define these structural ROIs from the desired residues, coordinates or even previous ligands.The user would be able to do so using the “Define Structural ROI” protocol in Scipion-chem.
- Map structural from sequence ROIs: The user would also be able to map some ROI from the protein sequence to its structure using the “Map sequence ROIs” protocol in Scipion-chem.For example, one might be interested in mapping conserved regions of the sequence onto the structure
- Predict binding pocket sites: Typically, ligand bind to protein pockets or cavities.We included several plugins including programs which predict the most promising pockets in the protein structure, based on variable factors. These are shown below.
3.1) scipion-chem-fpocket
FPocket is one of the most widely known programs for protein pocket detection. In this plugin we include the main tool for pocket prediction, FPocket, together with the tool for predicting pockets over a Molecular Dynamics simulation, MDPocket (in devel).
3.2) scipion-chem-p2rank
P2Rank is a ligand binding site predictor based on machine learning. In this plugin we include its main functionality for protein pocket prediction.
4) Ligand based approaches
This section is mainly included in the Scipion-chem core, and mainly consists on several rdkit functionalities that characterize small molecules which would later be used for docking. They are inspired on some of the TechOpenCADD talktorials
We included filters based on ADME or PAINS qualities; based on shape or fingerprint distance against a target ligand; and finally tools for generating, managing and filtering by pharmacophores.
5) Docking
5.1) scipion-chem-autodock
One of the main plugins in the Scipion-chem framework is the one for AutoDock. Here, we include the main docking functionalities of AutoDock4 and Vina, together with AutoLigand for protein pocket prediction and two protocols for the preparation of the receptor and the ligands (Meeko). At the moment of this writing, we have just included the AutoDockGPU and the Vina-1.2 versions from the ForliLab (https://forlilab.org/)
Scipion-chem-autodock automatically manages the necessary pdbqt format files and interconverts them from and to other file formats when necessary using openbabel or AutoDockTools when necessary. This way, the interoperability with other VDS tools can be achieved.
5.2) scipion-chem-lephar
With this plugin from Lephar, we include yet another tool for docking, LeDock. Together with the main docking tool, we include another protocol for target preparation using LePro.
5.3) scipion-chem-rosetta
Rosetta is widely known as one of the main software for molecular modelling. Even though this offers a lot of useful functionalities, as for today only protocols related to DARC docking are integrated. This includes a protocol for the target preparation and the protocol for DARC docking Regarding EM and protein modelling, this plugin also includes a protocol to fit a protein reference structure into an electron density map.
As for the rest of the plugins, the Scipion team is open to hear any suggestion or accept any help for integrating new functionalities, so let us know if you have any.
Also, user must know that the installation of Rosetta is not automatic. Instruction for a proper installation can be found in the scipion-chem-rosetta repo.
6) Molecular Dynamics
Molecular Dynamics (MD) simulate the state and movement of some molecules through time by modelling the energy interactions of their atoms and calculating the coordinates and velocities of each of them during a fixed number of steps. This modelling is never straight forward, and there are many parameters that must be tuned, but if it’s done properly one might get an accurate enough representation of the molecule in the real environment.
In Scipion-chem, we have included several programs that perform MD to model and simulate atomic structures (usually proteins) and even protein-ligand complexes.
In the VDS context, once you have some promising molecules docked to your receptor, one might want to do further computational studies to check whether the predicted interaction will be maintained in time or not. This can be achieved using molecular dynamics on the receptor-ligand complex.
6.1) scipion-chem-amber
Amber is a suite of biomolecular simulation programs. It contains open software for preparing and running MD simulations (AmberTools) and a second part which requires a license (Amber). By the time this documentation is writen, only AmberTools is integrated in Scipion-chem, so it can be installed automatically.
AmberTools also include cpptraj, a tool for managing and analyzing the resulting MD trajectories and is also being integrated in Scipion-chem-amber.
6.2) scipion-chem-gromacs
Gromacs A free and open-source software suite for high-performance MD and output analysis. By the time this documentation is writen, only the functionality for protein simulations is integrated. The parametrization of small molecules for simulating complexes will be handled and incorporated soon.
Different Gromacs tools are also included to manage and analyze the resulting trajectories.
6.3) scipion-chem-pwchemSchrodinger
Schrodinger is a full platform itself devoted to drug discovery and materials design. From Scipion-chem, we have integrated some of its tools and adapted them to or workflow, managing their maestro format files. Among the utils integrated, we have receptor (prepwizard) and ligand preparation (LigPrep), binding pocket prediction (SiteMap), docking (Glide) and molecular dynamics (Desmond).
This way, the user can design a full workflow of VDS using Schrodinger from the Scipion framework and analyze its results from the incorporated viewers.
Finally, user must know that the installation of Schrodinger is not automatic, since it depends on a license. Instruction for a proper installation can be found in the scipion-chem-pwchemSchrodinger repo.
7) Others
Further functionalities outside the most common VDS workflow are included in Scipion-chem. They include:
7.1) scipion-chem-modeller
Modeller is a tool used for protein homology modelling. In this plugin, we include a protocol for performing the typical homology modelling based on the sequence alignment of reference structures; and a protocol for modelling the structural change of mutations in the protein sequence.
7.2) scipion-chem-blast
Blast is the NCBI widely known program for searching biological sequences. In this plugin, we include tools for managing and configuring blast searchs, creating local BLAST databases and easily downloading NCBI data (protein/nucleotide sequences or small molecules) from their IDs